|
Özet:
GİRİŞ
Histidin Triad Nucleotide Binding Protein 1’i kodlayan HINT1 geni tumor supresyonu, tumor hücrelerinde apoptotic süreçlerin regülasyonu ve intraselüler sinyal iletiminde rol alır. HINT1deki resesif mutasyonlar, erken başlangıçlı, nöromiyotoninin eşlik ettiği motor aksonal periferik nöropatiye neden olur. Burada nadir bir homozigot missense c.355>T p. (Arg119 Trp) varyantının ortaya çıktığı moleküler genetik testlerle doğrulanan HINT1 olguları sunulmuştur.
OLGU-1
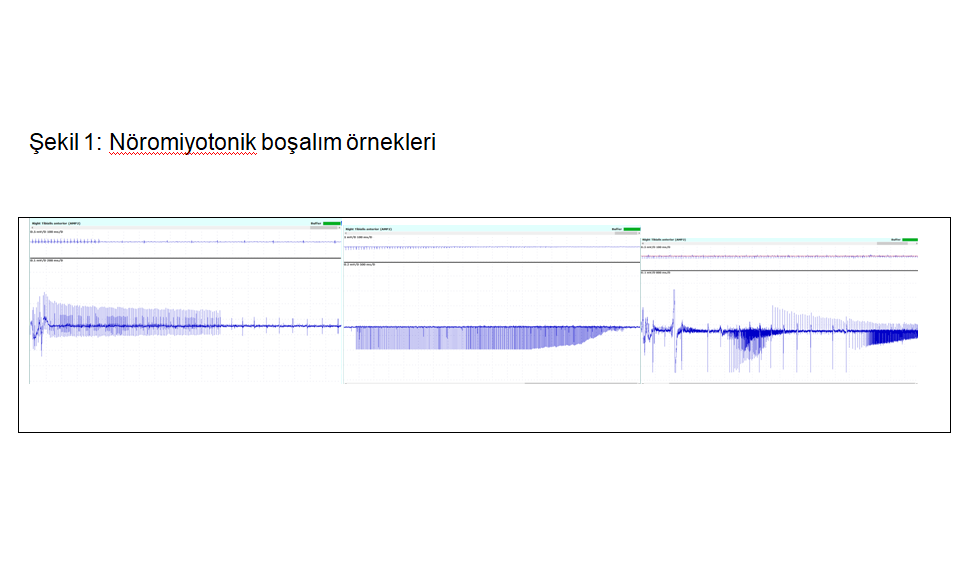
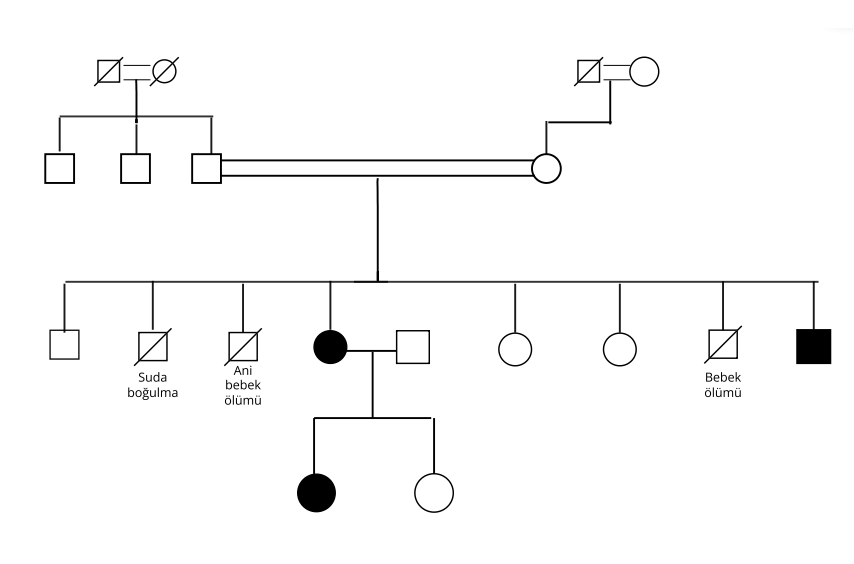
40 yaş kadın hasta 14 yaşında fark edilen yürüme güçlüğü, düşük ayak, düşme atakları, tremor yakınmasıyla başvurdu. Nörolojik muayenede alt ekstremitede baskın distal kas atrofisi, düşük ayak, postüral tremor ve el-ayak deformiteleri mevcuttu. DTR’ler hipoaktif olup duyu muayenesi normaldi. Spastisite veya diğer pyramidal bulgular yoktu. Serum CK düzeyi normaldi. Elektrodiagnostik incelemede duysal ileti incelemeleri normaldi. Motor ileti incelemerinde sağ peroneal sinir motor yanıt alınamadı,sağ ulnar ve sağ tibial sinir BKAP amplitude düşük saptandı. İğne elektromiyografisinde nöromiyotonik boşalımlar gözlendi (şekil-1). Distal kaslarda belirgin nörojenik patternde MUP değişiklikleri gözlendi. Nöromiyotoninin eşlik ettiği motor aksonal polinöropati sendromu ile uyumluydu. Birinci derece anne-baba akrabalığı olan olgunun erkek kardeşinde benzer hastalık öyküsü mevcuttu (Şekil-2).
OLGU-2
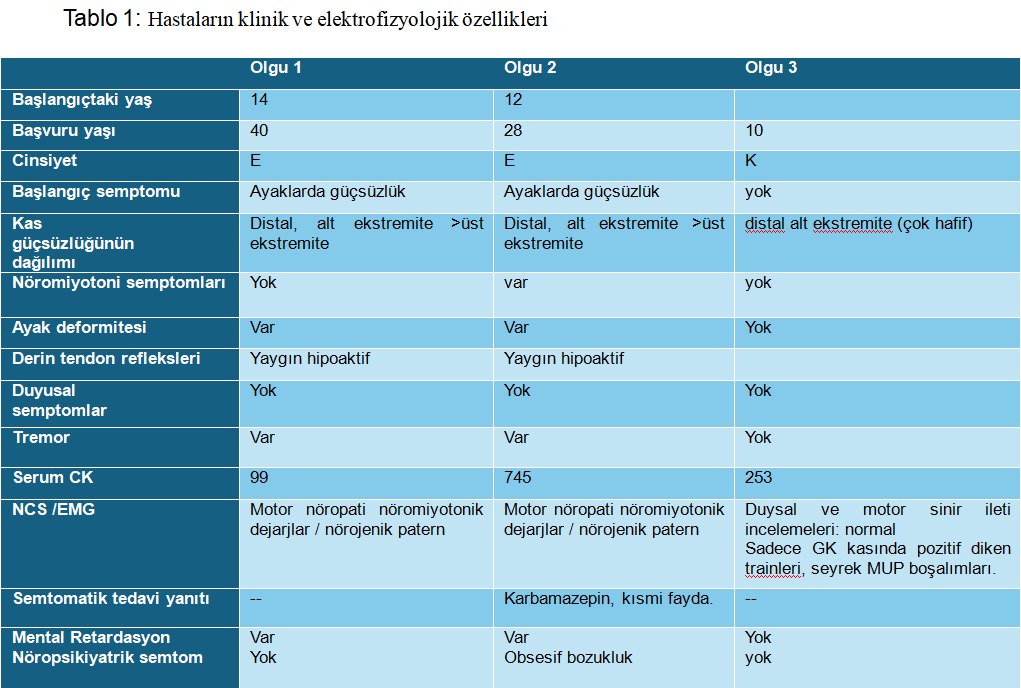
28 yaşındaki erkek kardeş, 12 yaşlarında başlayan, yavaş ilerleyen yürüme güçlüğü, kramp tariflemekteydi. Nörolojik muayenesinde distal kas atrofisi, alt ekstremitelerde belirgin distal kas zaafı, hiporefleksi, postüral tremor saptandı. Laboratuvar incelemelerinde CK:745 IU saptandı. Dış merkezde istenen SMN analizi negatifti. Elektrofizyolojik çalışmalar nöromiyotonik deşarjların eşlik ettiği motor nöronopatiyi gösterdi. Her iki hastaya yapılan yeni nesil dizileme (NGS) testinde HINT 1 (NM_005340.7): c.355>T p. (Arg119 Trp) varyantı homozigot formda mutasyon saptandı. Olgu-1’in asemptomatik olan 10 yaşındaki kızında ise heterozigot formda mutasyon saptandı. Klinik izleme alındı. Üç olgunun klinik ve elektrofizyolojik özellikleri tablo 1’de özetlenmiştir
TARTIŞMA
Bu vaka raporu yaşamın ilk veya ikinci on yılında ortaya çıkan nöromiyotoni ile birlikte motor aksonal polinöropatinin ayırıcı tanısında HINT1 ile ilişkili periferik nöropatinin dikkate alınması gerektiğini göstermektedir.
|
